
X. Shirley Liu received a Ph.D. in Biomedical Informatics and a Ph.D. minor in Computer Science from Stanford University in 2002. She went on to become a Professor with the Department of Biostatistics and Computational Biology at the Dana-Farber Cancer Institute and Harvard T.H. Chan School of Public Health. As a computational biologist, Shirley applied her expertise in cancer epigenetics and cancer immunology. Her work focused on algorithm development and data integration modeling for translational cancer research. Her laboratory developed widely used algorithms and tools for transcription factor motif finding, ChIP-chip/seq, chromatin accessibility profiles, CRISPR screen analyses, and tumor immune characterization. She left the Dana-Farber/Harvard in February of 2022 to become the CEO of GV20 Therapeutics.
Research interests
Our research focuses on algorithm development and integrative mining from high-throughput data to understand gene regulation in cancer biology. We have developed a number of widely used algorithms for transcription factor motif finding, ChIP-chip / ChIP-seq / DNase-seq / CRISPR Screen data analysis. Through integrating genome-wide transcription factor binding, chromatin dynamics, gene expression profiles, and chemical and functional screens, we try to model the specificity and function of transcription factors, chromatin regulators, RNA binding proteins, kinases, and lncRNAs in tumor development, progression, drug response and resistance.
Cancer Epigenetics
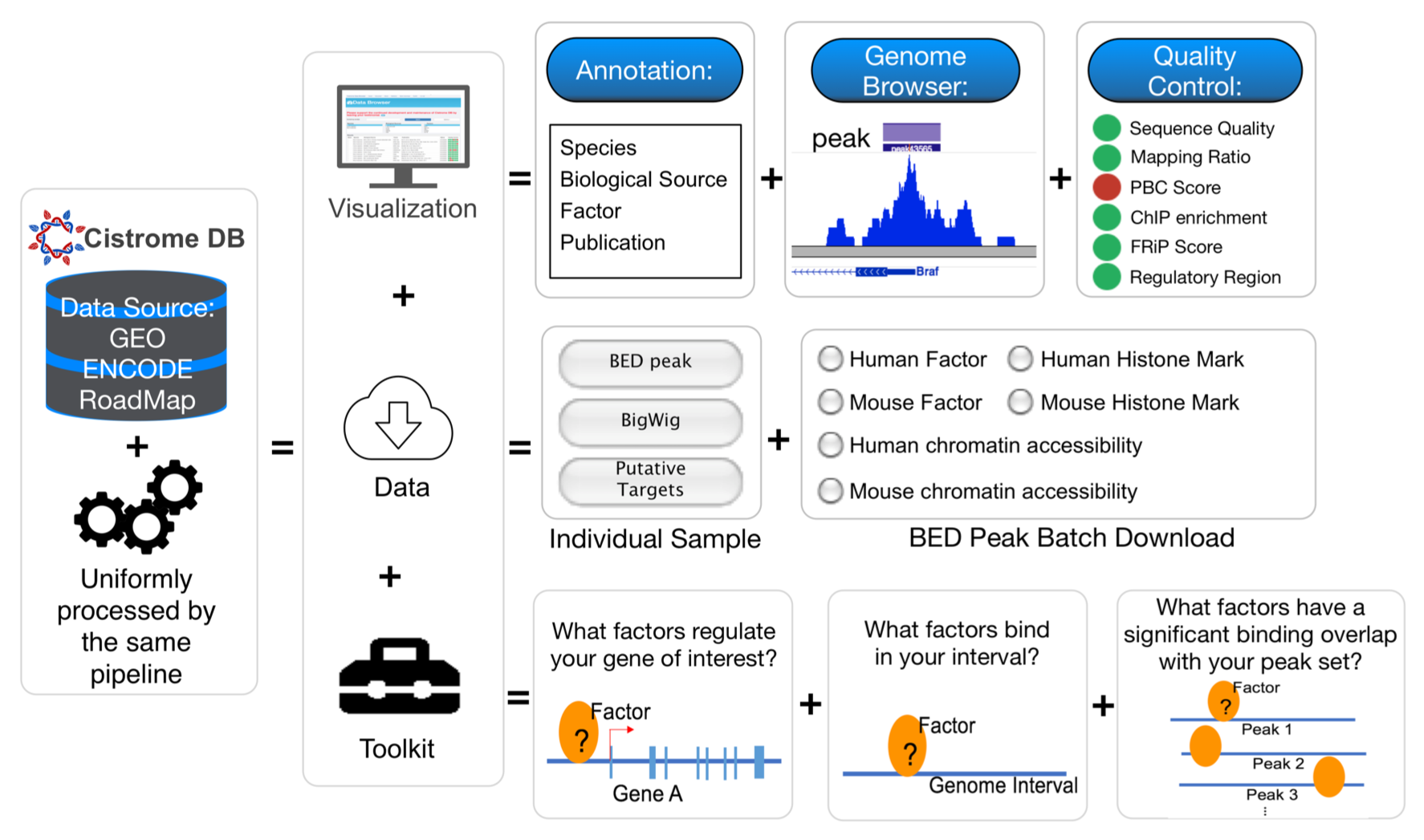
We have been developing algorithms (MACS, Cistrome, NPS, BETA) to facilitate the analysis of epigenomic data and use integrative modeling approaches to study genomic transcriptional and epigenetic gene regulatory mechanisms underlying tumorigenesis and progression. We are developing new methods to utilize the abundant public ChIP-seq data to infer functional enhancers that regulate the expression, and understand the targets and specificity of epigenetic drugs in cancers.
CRISPR screens
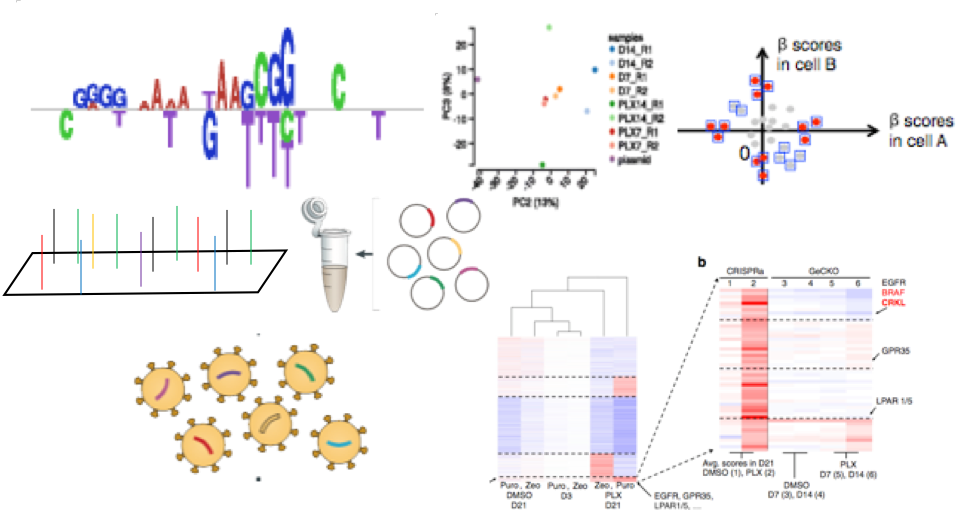
We are developing the computational methods for the design (SSC), analysis (MAGeCK), hit prioritization (NEST), and visualization (VISPR) of genome-wide CRISPR screens. We are also using this technology to identify key genes in breast and prostate tumor progression and drug resistance. We also develop CRISPR screen platforms to understand the functions of enhancers and long-noncoding RNAs, and identify synthetic lethal gene pairs in cancer that leads to optimized cancer precision medicine.
Cancer Immunology
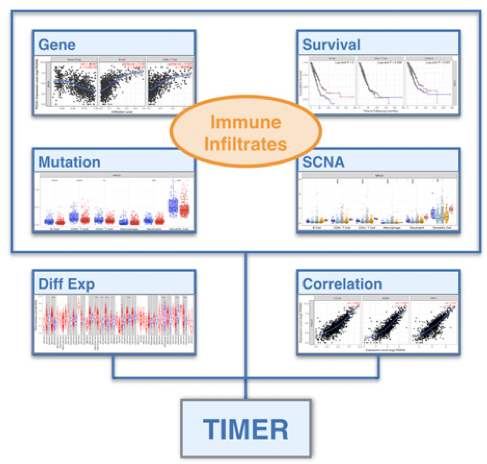
We are developing novel computational algorithms to systematically deconvolve the abundance of different immune cell types. Applying the deconvolution method on over 10K tumor samples from TCGA identified widespread clinical associations with tumor infiltrating immune cells. Our investigations on cancer vaccine and checkpoint blockade targets suggest that systematic analyses of tumor immunity have the potential to inform effective cancer immunotherapies.
Single Cell
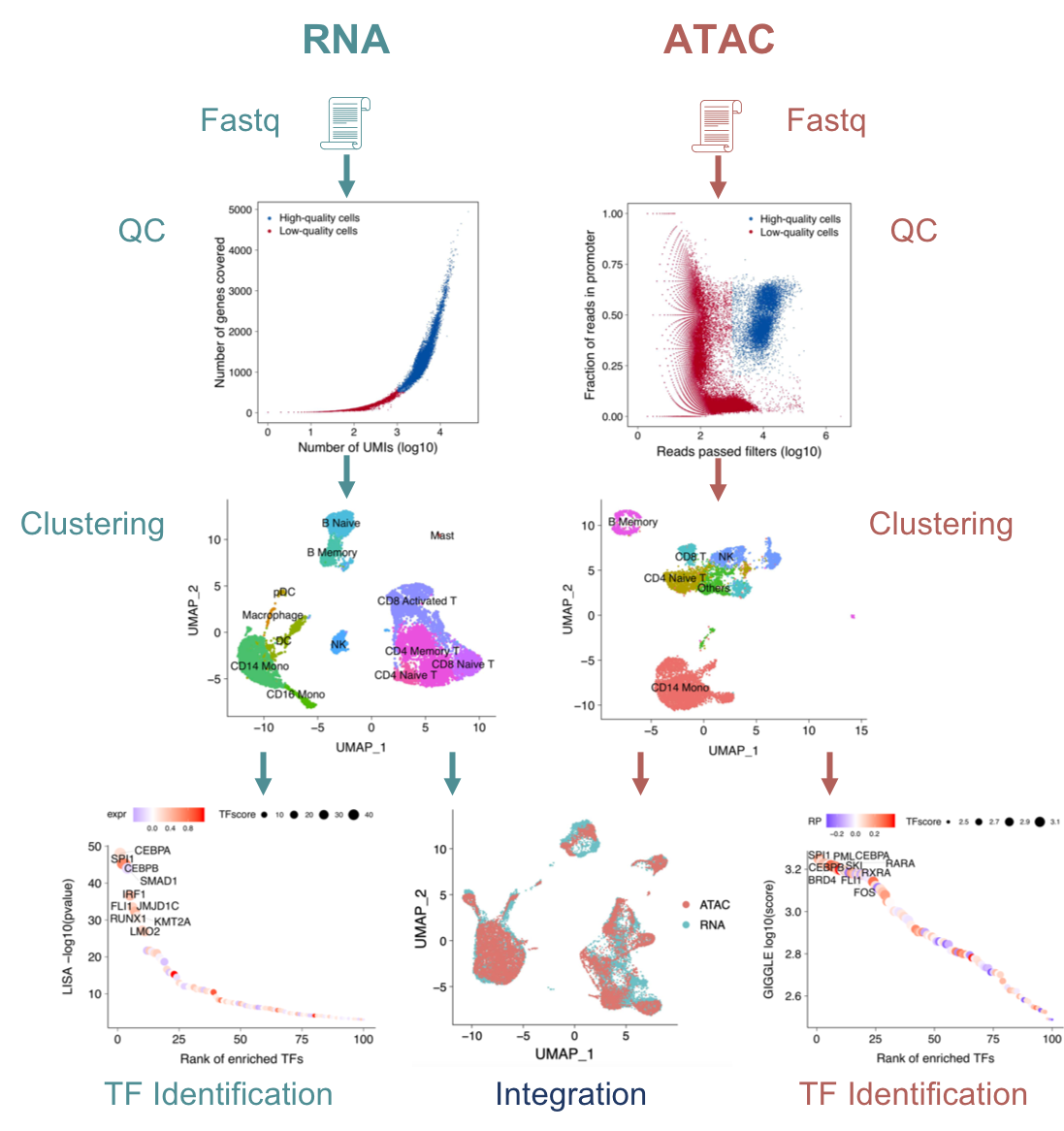
We are developing computational methods (MAESTRO) for integrative analysis of single-cell RNA-seq (scRNA-seq) and ATAC-seq (scATAC-seq) data. By integrating the public transcription factor (TF) ChIP-seq datasets in Cistrome DB, we are unraveling the regulatory network with tumor microenvironment and identifying key regulators between different immune cells. We are also developing novel computational algorithms for spatial transcriptomic data analysis.